GLM101 is in Phase 2 Clinical
Trials for PMM2-CDG
(MAY 2025)
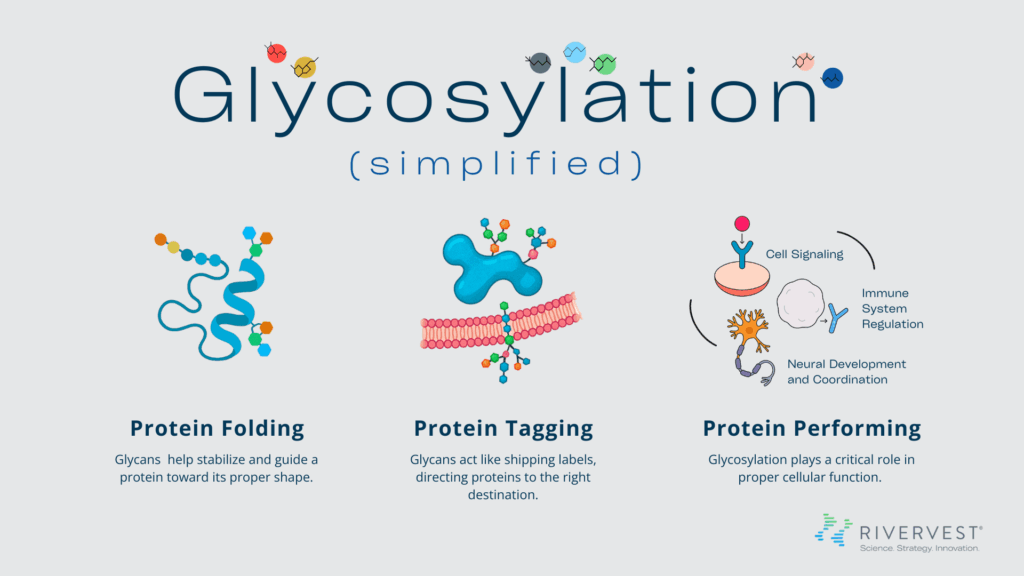
The human body is a vast and intricate network of biological systems and processes in which countless molecular interactions work in coordination to maintain health and respond to disease. When something goes wrong with one of these processes, it can be catastrophic to the system and life-altering for the person. Disruptions in glycosylation, for example, can lead to life-threatening disorders that can be maddeningly difficult to diagnose and even harder to treat.
In recognition of World CDG Day, RiverVest is spotlighting the critical role of glycosylation in human health, what happens when it’s disrupted, and what a novel sugar replacement therapy now in clinical trials could mean for patients with a rare congenital disorder of glycosylation — PMM2-CDG.